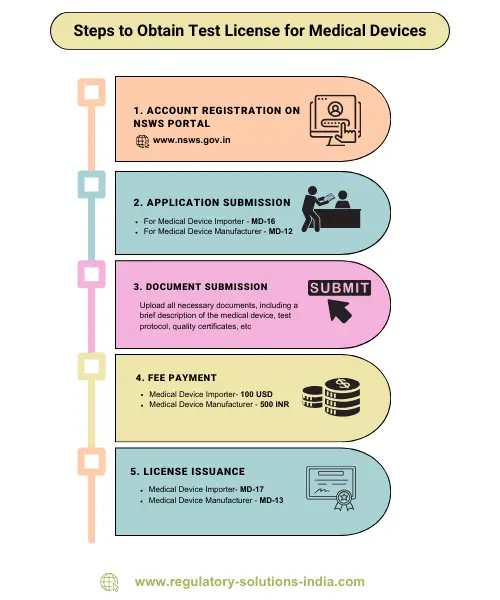
The Drugs Controller General of India (DCGI) has called for the timely reporting of adverse events related to medical devices by issuing a circular dated May 15, 2024. The move aims to strengthen the existing Materiovigilance Programme of India (MvPI) ensuring that medical devices are safe and reliable once in the market.
What does the Circular state?
The Materiovigilance Programme of India (MvPI) is an important programme for reporting adverse events, coordinated analysis related to medical devices, including in-vitro diagnostic devices, therefore it is suggested that all the license holders should also use the MvPI platform to report any adverse events/serious adverse events associated with the devices to enhance the procedure of identifying risk associated with medical devices.
What is MvPI?
The Materiovigilance Programme of India (MvPI) was launched by the Ministry of Health and Family Welfare, Government of India, on July 6, 2015, with the objective to monitor, record and analyse the root cause of adverse events or risks associated with the use of medical devices, including in-vitro diagnostics by healthcare professionals or patients/users and suggesting regulatory bodies to take appropriate action to improve Indian patients’ safety. Indian Pharmacopeia Commission (IPC) is functioning as National Coordination Center (NCC) for MvPI.
Objectives of MvPI:
The core objectives of MvPI are as follows:
- To promote the reporting of adverse events related to medical devices by clinicians, biomedical engineers/clinical engineers, hospital technology managers, pharmacists, nurses, technicians, medical-device manufacturers.
- To collect adverse event reports, assess them, and submit medical device reports to the medical device regulator.
- Voluntary registration of medical device manufacturers to:
- Report adverse events, associated.
- Root cause analysis and corrective/prevention action to IPC-NCC.
- To develop and implement an electronic reporting system (e-reporting).
- To support the health system where in procurement of medical device is only undertaken after studying adverse events associated with the medical device intended for procurement.
- To make it mandatory, in the long term, – for all healthcare providers under the Clinical Establishment Act to report adverse events associated with medical devices.
Important Responsibilities under MvPI:
Medical device organisations, including in-vitro diagnostics, are important stakeholders to make the MvPI programme a success. The key responsibilities of such organisations as a result of the MvPI are as follows:
- Robust Post-Market Surveillance: Post-market surveillance refers to the vigilance on the use of medical devices, including the information on the quality, safety. or performance of medical devices after the medical device is placed on the market. With a robust post-market surveillance system, organizations can reduce, -the chances of adverse events that may lead to any harm/ injury to the patient. Moreover, the regular vigilance process reduces the likelihood of the same type of adverse incident being repeated in different places at different times.
- Reporting of Medical Device Adverse Event: In case an adverse event or incident related to a medical device has been noticed by the manufacturer or healthcare service provider, or Medical Device Adverse Event Monitoring Centers (of public/private hospitals), it should be reported immediately, and its root cause of failure should be investigated and intimated to IPC-NCC.
- Reporting of Field Safety Corrective Action: A field safety corrective action (FSCA) notification form has been developed to notify the regulatory authority and the consignees regarding any corrective action or recall that has been initiated by the manufacturer/importer to reduce any serious adverse reaction associated with the use of medical devices. Concerned organizations must take cognizance of this form and use it for reporting their FSCAs.

Benefits of MvPI:
- It is crucial for providing secured, sensible and responsible healthcare to citizens in India.
- It contributes to establishing a system in India for systematic, scientific and practical means of screening large medical device adverse events datasets at the national level.
- It contributes to public health by identifying potential safety issues more accurately and quickly.
- It promotes patient safety by identifying adverse events that could result in patient harm.
- It encourages Medical Device Manufacturers/ Importers to put products in market with a sense of ethical business, analyze and improve design and performance of products.
- It can provide supportive data to improvise product standards developed by ISO/BIS.
Conclusion:
MvPI is a critical components of India's healthcare delivery system, guaranteeing the safety and efficacy of medical devices while increasing healthcare quality. Prioritizing transparency and employing cutting edge technology can create an efficient Materiovigilance system which can protect public health while improving healthcare quality.
For more information on India's Materiovigilance Programme and its effect on medical device safety, visit, https://www.ipc.gov.in/mandates/materiovigilance-programme-of-india-mvpi/about-us.html.
How can Regulatory Solutions India Help You?
Regulatory Solutions India (RSI) is your reliable partner when it comes to medical device regulation. Since 2011, they have successfully registered over 450 products ranging from medical devices, IVDs and cosmetics across 25+ categories such as stents, catheters, intraocular lenses, orthopedic implants, ablation devices surgical dressings, hypodermic syringes/needles and more from clients from 15+ countries.
RSI provides its clients with a unique combination of technical, strategic and project management support backed by rich industry experience. We help companies register their products with India's central licensing authority (CDSCO) while offering expert knowledge into CDSCO medical device registration and import license processes in order to navigate successfully the regulatory landscape.
Why Choose RSI?
- Regulatory Strategy for Medical devices: Analyse the portfolio, interpret regulatory requirements, identify potential problems, and design the right strategy for medical devices accordingly.
- Regulatory Application Submission to CDSCO: Review regulatory documents, validate dossiers and submit applications for Import License.
- Registration Approval: Technical support for scientific meetings with regulators and for responding to regulatory queries.
- Post Registration Support: It also offers post registration support in labeling recommendations & regulatory compliance, post registration compliance reporting obligations and advise on impact, if any, due to regulatory changes.
Take Action Today
Partner with Regulatory Solutions India to ensure the safety and security of the medical devices. Visit us here.